log Kow
The ratio of the concentration of a solute between water and octanol is a well-known property that is commonly used as a measure of hydrophobicity. The ratio is essentially independent of concentration, and is usually expressed in logarithmic terms (log KOW or log Poct), which are better suited for use as a free-energy based parameter in thermodynamic equations. There are a significant number of estimation methods that have been developed for log KOW, and while no single review could possibly cover them all, the review by (Mannhold et al, 2009) provides a comprehensive summary of about 35 different methods. This report focuses primarily on the more commonly used methods, identified in Table 8 above, as KOWWIN, ACD/LogP, SPARC, and ALogPS v. 2.1.
KOWWIN
KOWWIN is an estimation method included in the suite of programs known as EPI (Estimation Programs Interface) Suite™. Other estimation methods included in EPISuite allow the user to estimate the properties for Henrys Law Constant, water solubility, vapour pressure, melting point, boiling point, etc., as well as estimates of a number of environmental fate endpoints (e.g. bioconcentration factor, atmospheric abiotic degradation etc.). It was developed and is maintained by the US Environmental Protection Agency (EPA) and the Syracuse Research Corporation (SRC). The current version of the program (version 4.1) is freely available for download at http://www.epa.gov/opptintr/exposure/pubs/episuite.htm. To estimate property and environmental fate endpoints a user simply inputs the chemical of interest to the interface as a SMILES code or by CAS, and all estimation programs can be run together. Alternatively, the user can run each of the estimation methods as stand-alone programs. Furthermore, a database is implemented containing one or more physical-chemical properties for more than 40,000 substances. These data may refine some of the model calculations.
The KOWWIN program estimates KOW using an atom/fragment contribution approach. It was developed by Meylan and Howard (Meylan and Howard, 1995; Meylan et al, 1996; Meylan and Howard, 2000) using a training set of 2473 compounds, and a validation set of 10589 compounds representing molecules with simple to complex structures. From this 150 atom/fragments were defined and are used in combination with 250 correction factors, which account for steric interactions, H-bonding, and effects from polar substructures. The following general equation is used by KOWWIN for estimating log KOW:

Where fi is the fragment coefficient, ni the frequency of the fragment in the structure, cj the correction factor coefficient, nj the frequency of the factor in the structure, and 0.229 is the constant value generated by the multiple linear regression. KOWWIN also provides an alternative approach, whereby an experimental adjusted value can be used to better estimate the log KOW of an unknown molecular structure based on the measured value of a molecule with a similar structure. Regarding calculations for charged molecules the most recent version of EpiSuite offers the possibility to draw the molecule including charges of single atoms.
ACD/logP
Like KOWWIN, the ACD/logP estimation method uses a fragment-based approach, whereby the contributions of separate atoms, structural fragments, and intramolecular interactions between different fragments are considered (Petrauskas and Kolovanov, 2000). The individual contributions have been quantified based on a database of about 18,400 structures having experimental log KOW data, which resulted in over 1,200 different functional groups being defined with fragment contributions. The database for intramolecular interaction contributions contains increments for over 2400 different types of pair-wise group interactions. If a fragment or intramolecular interaction contribution for a molecule that is being estimated are not found, then the ACD/LogP program will calculate it through the use of a special secondary algorithm, and a larger uncertainty against the log KOW estimate is defined. The value of log KOW is thus calculated as:
![]()
Where fi is the fragmental increments, Qj are the increments associated with superfragments, Fijk are increments of interactions between any two (ith and jth) groups separated by k-number of aliphatic, vinylic, or aromatic atoms.
The ACD/logP is part of a commercially available software package, licensed by Advanced Chemistry Development. More information is available at http://www.acdlabs.com/.
SPARC
SPARC (SPARC Performs Automated Reasoning in Chemistry) is a co-project founded by the Environmental Research Laboratory of the US-EPA and the Department of Chemistry of the University of Georgia. It uses existing knowledge to estimate different physical-chemical properties of substances as well as chemical reactivity parameters. On the basis of existing mathematical models SPARC is mainly working on theory and mechanism of oriented substructure. Its computational approach is to analyse a substance's structure as well as its sub-structures relative to its different reactivities. Common theories in organic chemistry are applied to predict intermolecular interactions based on all available interaction forces e.g. dipole moment, induction, H-bonding etc. by use of the molecular sub-structure of the molecule. These sub-groups are functional groups with an intrinsic reactivity.
Within the current version 4.5 of SPARC estimations can be made regarding pKa (also for solids and gases), log D, Henry's Law Constant as a function of pH, hydrolysis etc. as well as different properties like vapour pressure, boiling point, electron affinity, density, polarisability, Henry's Law Constant, solubility in water and other media, and distribution coefficients (e.g. log KOW).
The fundamental approach used by SPARC is based on a blending of linear free energy relationships (LFERs), structure activity relationships, and perturbed molecular orbitals (PMO) to describe a variety of physical and reactivity parameters (Hilal et al., 2004). SPARC describes intermolecular interactions as a summation of all free energy changes :
![]()
Where the first four terms describe the change in the intermolecular free energy interactions accompanying the physical process of interest, in this instance the distribution of a chemical between octanol and water. Unlike the fragment-based approaches which calculate log KOW based on the contributions of fragments contained within a molecule, SPARC calculates log KOW by first calculating the activities of the chemical at infinite dilution in both octanol and water:
![]()
Where ƒÁ‡ are the activities at infinite dilution of the chemical in each phase, and Rm = -0.82, which is a coefficient that converts the mole fraction concentration to moles/l for water and octanol saturated with water. It is suggested that the water in the octanol phase makes the approach more realistic, as opposed to assuming a edry' octanol phase, since experimental methods will be using a saturated octanol phase in the test system. Although it is noted that the importance of including water in the octanol phase has a bigger influence on larger, hydrophobic molecules, as opposed to smaller molecules (Hilal et al. 2004).
The estimation method is freely available at http://archemcalc.com/sparc. The developers have also recently made available a commercial version that allows users to operate a stand-alone application, which may be appealing where proprietary chemicals are being assessed, or where batch runs are desirable.
Both SPARC and ACD/logP methods allow for the estimation of log DOW values, whereby the influence of pH on ionisable organic compounds is accounted for in the calculations. The availability of this functionality has recently been utilised by Rayne and Forest (2010), who estimated the log DOW values of 543 ionisable organic compounds listed on the Canadian Domestic Substances list. The study by Rayne and Forest (2010) demonstrated how the use of DOW can significantly influence how chemicals might be screened for bioaccumulation potential and long-range atmospheric transport, as opposed to relying on log KOW values, and they suggest that the use of log DOW would be a more appropriate metric. This argument is echoed by Wells (2006) who also suggest that the use of log DOW would lead to improvements in understanding the fate of ionisable organic compounds within waste water treatment systems. Consequently, the performance of predictions of log DOW is explored for both SPARC and ACD/LogP against measured data assembled in the dataset reported in Appendix F. The use of log DOW within modelling tools is further assessed in Chapter 4, where the Task Force also speculate on its use within the regulatory framework as a potential trigger value for bioaccumulation potential and persistence testing.
ALogPS v. 2.1
Using 75 descriptors, the ALogPS v. 2.1, freely available at http://www.vcclab.org/, uses a neural network method, based on 12908 molecules, with a RMSE of 0.35 (Tetko and Tanchuk, 2002; Tetko, 2002). The method used in ALogPS is based on E-state indices, which were developed to cover both topological and valence states of atoms, and have been used to develop QSARs for a number of physical-chemical and biological properties (Mannhold et al, 2009). The Task Force has included this method in their review based on results reported by Mannhold et al (2009), where the method is shown to perform relatively well on an extensive dataset. Indeed the results reported by Mannhold et al (2009) suggest that of the methods reviewed by the Task Force, the ACD/LogP performs best, followed by ALogPS and KOWWIN, both of which show similar performance, and then SPARC, which performed more poorly than the other three methods. Consequently, the four methods included in this review should thus reflect a range of relative performance with respect to estimating log KOW the ratio of the concentration of a solute between water and octanol is a well-known property that is commonly used as a measure of hydrophobicity. The ratio is essentially independent of concentration, and is usually expressed in logarithmic terms (log KOW or log Poct), which are better suited for use as a free-energy based parameter in thermodynamic equations. There are a significant number of estimation methods that have been developed for log KOW, and while no single review could possibly cover them all, the review by Mannhold et al (2009) provides a comprehensive summary of about 35 different methods. This report focuses primarily on the more commonly used methods, identified in Table 8, as KOWWIN, ACD/LogP, SPARC, and ALogPS v. 2.1.
Comparison of log KOW estimation methods
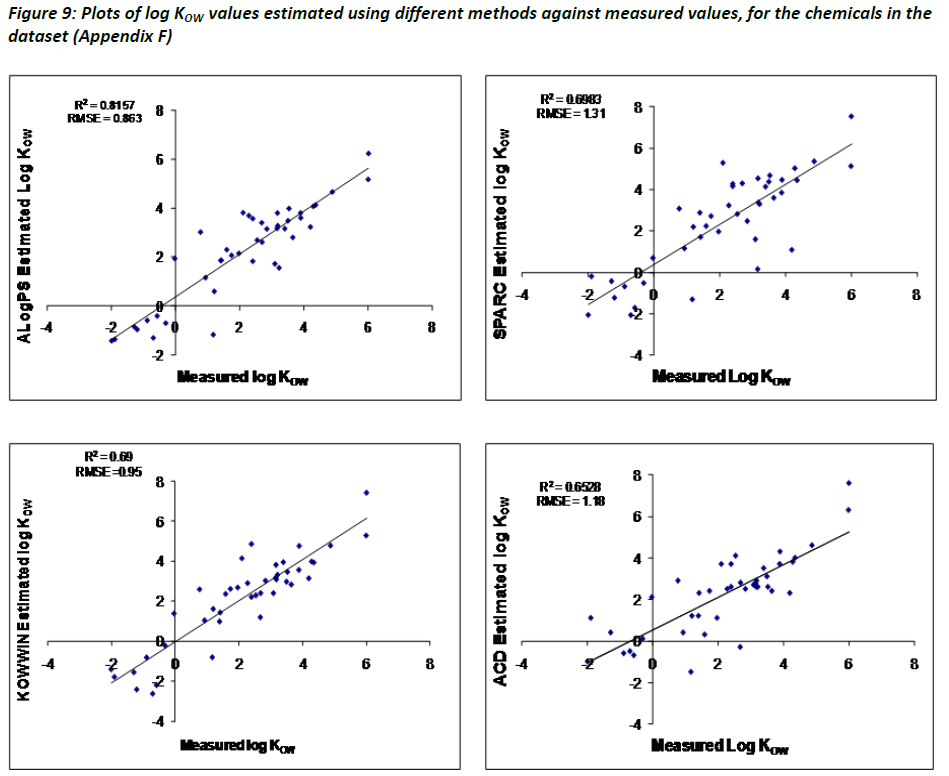
Figure 9 summarises the comparison of the log KOW estimated using the four different methods referred to in Table 8 against measured data reported in Appendix F. Results for SPARC and KOWWIN were shown to be comparable to one another, with ALogPS performing slightly better. ACD/LogP, which performed better than the other three methods in the review by Mannhold et al (2009), has a root-mean-square error (RMSE) of 1.18, which is comparable to previously reported observations.
Based on the results presented in Figure 9 for the class of chemicals investigated in this study, it is generally noted that ALogPS performs better than KOWWIN, ACD/LogP, and SPARC in estimating log KOW. This is a curious observation, particularly given the near ubiquitous use of KOWWIN within regulatory agencies for screening chemicals for BCF and persistence testing. Careful interpretation of the data by the TF suggests that caution be used when relying on any single estimation program as a tool for estimating such trigger values, especially where molecules with complex molecular structures are being assessed.